
Fenylketonuri er en arvelig metabolsk sykdom. Den består i en feil transformasjon av en av aminosyrene - fenylalanin. Et riktig kosthold er veldig viktig i behandlingen av fenylketonuri. Finn ut hva som er definisjonen av fenylketonuri og hva er dens symptomer.
Innholdsfortegnelse:
- Fenylketonuri - definisjon
- Fenylketonuri - behandling
- Fenylketonuri - diett
- Fenylketonuri - forbudet mot fenylalanin
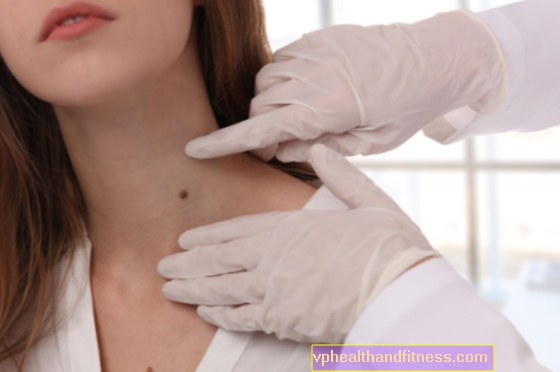
Fenylketonuri er en arvelig sykdom, noe som betyr at du ikke kan fange den. Personer med fenylketonuri kan avgi en karakteristisk "mousy" lukt. Et barn kan bli født syk når både far og mor lider av denne sykdommen eller bærer den. Sannsynligheten for at dette vil skje er 25 prosent. Det samme er sjansene for at et barn blir født sunt, og en og en halv vil ikke lide av seg selv, men vil bli en bærer av sykdommen. Imidlertid, hvis bare en av foreldrene er berørt av PKU, vil barnet være sunt. For tiden kan fenylketonuri oppdages ved screening av nyfødte.
Hør om fenylketonuri, medfødt metabolsk sykdom, dens symptomer og behandling. Dette er materiale fra den LYTTENDE GODE syklusen. Podcaster med tips.
For å se denne videoen må du aktivere JavaScript, og vurdere å oppgradere til en nettleser som støtter -video
Fenylketonuri - definisjon
Fenylketonuri er medfødt og progresjonen er rask og irreversibel. Derfor er det ekstremt viktig å oppdage det så tidlig som mulig. Fenylketonuri er den unormale forandringen av en av aminosyrene fenylalanin. Det bygger seg opp for mye i kroppen og skader hjernen, og til og med forårsaker alvorlig mental retardasjon. Sykdommen er ledsaget av nevrologiske symptomer: kramper, alvorlig muskelspenning og skjelving og atferdsforstyrrelser.
I Polen og i mange andre land blir testene for denne sykdommen utsatt for barn på livets tredje dag, og testen gjentas etter to uker. Hvis sykdommen blir tydelig i løpet av de første ukene av livet og tilstrekkelig ernæring implementeres umiddelbart, vil babyen utvikle seg skikkelig.
Fenylketonuri - behandling
Det finnes ingen kur for det. Du kan bare forhindre kroppsforgiftning og hjerneskade ved å følge et passende, veldig strengt kosthold. Barnet må følge det til hjernen bremser utviklingen, dvs. til 12-14 år. Etter det kan kostholdsbegrensninger lett liberaliseres. Leger kommer imidlertid til den konklusjonen at en person som lider av fenylketonuri, bør følge mer eller mindre strengt en diett som er passende for denne sykdommen gjennom hele livet. Dette gjelder spesielt kvinner med fenylketonuri som blir gravide. Hvis de slutter å følge dietten tidligere, bør de gå tilbake til det i løpet av denne tiden. Økte nivåer av fenylalanin i blodet kan føre til at skadelige stoffer kommer inn i babyens blodomløp og forårsaker permanent skade på hjernen.
Fenylketonuri - diett
En streng diett må foreskrives av en lege spesialisert i behandling av fenylketonuri, som vil vurdere hvor mye fenylalanin trygt kan leveres til pasientens kropp. For at dette skal skje, er regelmessige og hyppige blodprøver av aminosyrenivået avgjørende. Vanligvis blir et barn under tre år testet ukentlig, deretter annenhver uke og månedlig når barnet fyller sju år.
Fenylketonuri - forbudet mot fenylalanin
Mat som forbrukes av barn med PKU, må bare inneholde små mengder fenylalanin (små mengder er nødvendige for riktig utvikling) - en aminosyre som kroppen ikke klarer å omdanne. Derfor bør du fjerne alle kjøttprodukter, fisk, egg, ost, melk og dets produkter, brød og mange andre melprodukter fra maten.
Manglene på næringsstoffene i disse produktene suppleres med spesielle preparater som inneholder de nødvendige aminosyrene, vitaminene og mineralsaltene. Detaljer må utarbeides av en ernæringsfysiolog for at måltidene skal være balansert og velsmakende på samme tid. Siden kostholdet til pasienter med fenylketonuri må endres avhengig av helsetilstanden og alderen til barnet, krever riktig bruk konstant og hyppig konsultasjon med lege og ernæringsfysiolog.
Anbefalt artikkel:
GENETISKE SYKDOMMER: årsaker, arv og diagnose













-przyczyny-objawy-leczenie.jpg)


