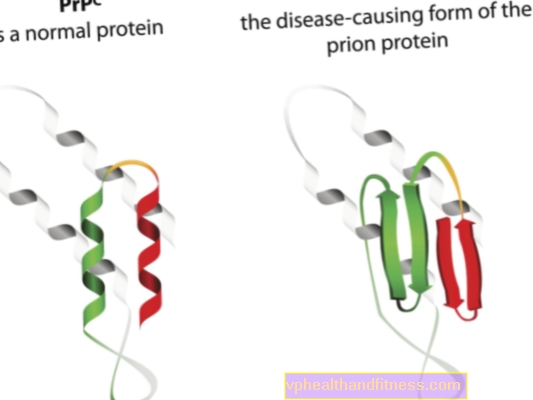
Spongiform encefalopati (prionsykdommer) er de sykdommene der patologiske former for prionproteiner er involvert i utviklingen. Vi vet mer og mer om prionsykdommer, men de viktigste aspektene er fremdeles ukjente - for tiden har medisin ikke midler til å kurere pasienter fra disse sykdommene.
Spongiforme encefalopatier, eller prionsykdommer, kan utvikle seg i løpet av livet, mens andre stammer fra arvelige genmutasjoner fra fødselen. Innenfor denne gruppen er det flere enheter som forekommer hos mennesker, eksempler er Creutzfeldt-Jakobs sykdom eller dødelig familiær søvnløshet.
Prionsykdommer har vært veldig mystiske i lang tid. I motsetning til andre patogener, som bakterier, virus eller sopp, inneholder de ikke nukleinsyre - prioner er bare laget av proteiner. Teorien om prionsykdommer ble oppdaget av S. Prusiner, denne oppdagelsen ble høyt verdsatt i det vitenskapelige samfunnet - i 1997 mottok forskeren Nobelprisen i medisin. Selv om det har gått relativt mange år siden prion-konseptet ble født, mener noen forskere fremdeles at det er ufullstendig og undersøker nærmere karakteren av disse forholdene - noen av faktorene som er ansvarlige for spongiform encefalopati er nå bekreftet.
Prionsykdommer: årsaker
Etiologien til prionsykdommer er relatert til transformasjonen av normale prionproteiner til patogene, patogene former. Prioner er proteinmolekyler som finnes i kroppen til alle. Deres funksjon er ikke helt klar ennå, men det er kjent at prionproteiner under normale forhold ikke skader kroppen. Imidlertid, når prioner endrer strukturen og blir patogene partikler, utvikler det seg en av flere spongiforme encefalopatier. Prioner som naturlig forekommer i kroppen blir referert til som PRPC, mens unormale former blir referert til som PRPSC. Sistnevnte er et alvorlig problem, ikke bare fordi de kan samle seg i nervevævet i form av avleiringer og generere skade på det, men også fordi de har evnen til å transformere normale prioner til en misdannet form (enkelt sagt, PRPSC kan "infisere" normale proteiner med dets patogene potensial).
Les også: Huntingtons sykdom (Huntingtons chorea): årsaker, symptomer, behandling Muskelskjelv - årsaker. Hva betyr muskel tremor? Sykdommer som dreper raskest: SHOCK, EBOLA, DAMN, ATTACK, EMERGENCY [GALE ...I utgangspunktet er det 3 årsaker til spongiform encefalopati:
- sporadisk (patogen mutasjon forekommer i somatiske celler, den oppstår i løpet av pasientens liv),
- familie (som følge av belastningen av mutasjoner arvet fra foreldrene),
- Passasje (relatert til innføring av patogene prioner i menneskekroppen, f.eks. Gjennom veksthormonpreparater forurenset med disse partiklene eller hornhinnetransplantasjon fra en person som lider av svampformet encefalopati).
Spongiform encefalopati: Creutzfeldt-Jakobs sykdom
Creutzfeldt-Jakobs sykdom (CJD) ble først beskrevet tidlig på 1920-tallet. Det er fire typer sykdommer:
- sporadisk CJD (den vanligste, står for opptil 9/10 av alle CJD-tilfeller)
- hjembyen CJD
- overveldet av CJD
- variant av CJD
Det kliniske bildet i løpet av forskjellige varianter av Creutzfeldt-Jakobs sykdom kan være variabelt. De vanligste plagene i løpet av denne gruppen spongiforme encefalopatier er:
- demenslidelser (inkludert progressiv forverring av hukommelse, oppmerksomhet og konsentrasjon)
- myoklonus (ufrivillige bevegelser som plutselige muskelrykk)
- cerebellar dysfunksjon (manifestert for eksempel av balanseforstyrrelser)
- tåkesyn
- pyramidale og ekstrapyramidale symptomer
I løpet av CJD-varianter kan også psykiske lidelser (f.eks. Angst, deprimert humør), smerte og andre ufrivillige bevegelser enn de som er nevnt ovenfor, dukke opp.
Prognosen for Creutzfeldt-Jakobs sykdom er dårlig - for eksempel, hos pasienter med sporadisk CJD, tar det i gjennomsnitt fire til fem måneder fra sykdomssymptomers begynnelse til døden.
Spongiform encefalopati: Gerstmann-Straussler-Scheinker syndrom
Gerstmann-Straussler-Scheinker syndrom (GSS) kjører vanligvis i familier og er forårsaket av en arvelig mutasjon i PRNP-genet. Det anses å være den langsomste utviklingen av spongiform encefalopati. GSS-teamet inkluderer:
- spinocerebellar ataksi
- dysartri
- demenslidelser
- svelgeforstyrrelser
- nystagmus
- økt muskelspenning
Pasienter diagnostisert med GSS har variabel tid, og hos noen pasienter oppstår døden mer enn 10 år etter utbruddet.
Spongiform encefalopati: dødelig familiær søvnløshet
Dødelig familiær søvnløshet er en prionsykdom forårsaket av mutasjoner i PRNP-genet. Sykdommen er ekstremt sjelden og har hittil blitt diagnostisert i 28 familier over hele verden. I løpet av dødelig familiær søvnløshet er det første symptomet manglende evne til å sove. Dette problemet resulterer i angstlidelser og pasienten opplever hallusinasjoner. Effekten av konstant mangel på natthvile er dysfunksjon i det autonome systemet (inkludert endringer i hjertefunksjon, svette og fordøyelsessystemforstyrrelser), det er også en progressiv reduksjon i kroppsvekt. I mer avanserte stadier av dødelig familiær søvnløshet vises hormonelle forstyrrelser, og symptomer på demens oppstår i løpet av sykdommen.
Prognosen for dødelig familiær søvnløshet, som for andre spongiforme encefalopatier, er dårlig: pasienter dør vanligvis innen tre år etter utbruddet.
Spongiform encefalopati: prionopati med variabel følsomhet for protease
Forekomsten av disse spongiforme encefalopatiene er hovedsakelig relatert til mutasjoner i PRNP-genet. Imidlertid gjelder disse mutasjonene forskjellige kodoner av dette genet, og det skilles derfor mellom flere forskjellige prionsykdommer. En relativt nylig beskrevet (i 2008) enhet er prionopati med variabel følsomhet for protease. Mennesker som lider av denne sykdommen bærer mutasjoner i så mange som tre kodoner av PRNP-genet.
Ved prionopati med variabel følsomhet for protease, opplever pasienter:
- kognitiv svikt
- ekstrem alvorlighetsgrad av psykiatriske lidelser: de kan være eufori og uro, men også betydelig apati
- dysartri
- afasi (språkforstyrrelser)
Den gjennomsnittlige varigheten av sykdommen i denne prionopati er mindre enn 4 år.
Spongiform encefalopati: kuru
Kuru regnes nå som en sykdom som praktisk talt ikke eksisterer lenger - den ble funnet hos representanter for stammer fra Papua Ny-Guinea, som praktiserte kannibalistisk oppførsel. Det dominerende symptomet på denne spongiform encefalopati er progressiv cerebellar ataksi. Det kan ledsages av ufrivillige bevegelser (hovedsakelig i form av chorea, skjelving og athetose), samt urin og fekal inkontinens. Pasienter på kuru opplever også betydelige humørsvingninger, de utvikler primitive reflekser (f.eks. Suging). Helt et karakteristisk problem i tilfelle av denne prionsykdommen er tvungne anfall av gråt eller latter - på grunn av det sistnevnte fenomenet blir kuru noen ganger referert til som "latterdøden".
Spongiform encefalopati: diagnose
Prionsykdommer kan mistenkes på grunnlag av pasientens symptomer. Imidlertid er de ganske uspesifikke, da de også kan vises i løpet av en rekke andre sykdommer som ikke er relatert til prioner. Av denne grunn brukes følgende også til diagnosen spongiform encefalopati:
- avbildningstester (f.eks. magnetisk resonansavbildning, som gjør det mulig å oppdage endringer relatert til degenerasjonen av hjernen av prionproteiner),
- laboratorietester (for eksempel vurdering av proteinkonsentrasjoner i cerebrospinalvæsken, f.eks. MAP-tau, S-100 eller 14-3-3 proteiner),
- genetiske tester (for å oppdage tilstedeværelsen av mutasjoner hos pasienten),
- immunhistokjemiske tester (ved bruk av antistoffer mot prionproteiner).
Diagnosen kan også bekreftes ved obduksjon av hjernen, der det er mulig å finne endringer som er karakteristiske for spongiforme encefalopatier. Disse kan være svampete lesjoner, forskjellig fordelt og med en annen struktur (avhengig av den spesifikke sykdomsenheten), amyloide plakk og nevronale defekter.
Spongiform encefalopati: behandling
Prionsykdommer er for tiden uhelbredelige - til tross for mange studier som har pågått i mange år, har medisin fortsatt ikke medisiner som kan bremse eller fullstendig hemme fremdriften deres. Symptomatisk behandling brukes hos pasienter med spongiform encefalopati, som er rettet mot å lindre intensiteten av symptomene og forbedre livskvaliteten så mye som mulig.
Arbeidet med behandling av spongiforme encefalopatier pågår imidlertid fortsatt. Forskere prøver å bruke forskjellige metoder - det første eksemplet er genterapi. De ville påvirke nukleinsyrer og mutasjonene som er tilstede i strukturen deres - formålet med å anvende genterapi ville være å nøytralisere feil i den genetiske koden. En annen tilnærming er grunnlaget for immunterapi - arbeidet pågår for å skape antistoffer hvis rolle er å eliminere patogene prioner. En annen metode som ser potensialet for å bekjempe spongiforme encefalopatier er behandling med bruk av syntetiserte proteinmolekyler, som en gang introdusert i pasientens kropp vil nøytralisere patologiske proteiner.
Anbefalt artikkel:
Encefalopatier - årsaker, typer og symptomer















-przyczyny-objawy-leczenie.jpg)


